
Registration for NGSchool 2025: Sequencing Toolbox for Computational Biologists is now closed. Thank you to all who took the time to answer the questions and solve the problems in the registration form. Here we publish the solutions.
Correct answers are marked in bold.
Bioinformatics / Biological Understanding
The first set of questions checked that the applicants understand the basics of biological study design, can critically interpret presented results and successfully utilize publicly available resources.
B1
You performed bulk transcriptomic sequencing of mouse blood samples. However, when you tried to align the reads to a reference genome, you found: 0% uniquely mapped reads, 10% multi-mapped reads, and 90% unmapped reads. Which of the following options cannot explain the failure in mapping:
A) Samples have been contaminated
B) Adapter trimming was not performed
C) You did not deplete the ribosomal RNA
D) You are aligning with the wrong reference genome
E) The sequencing quality was poor
B2
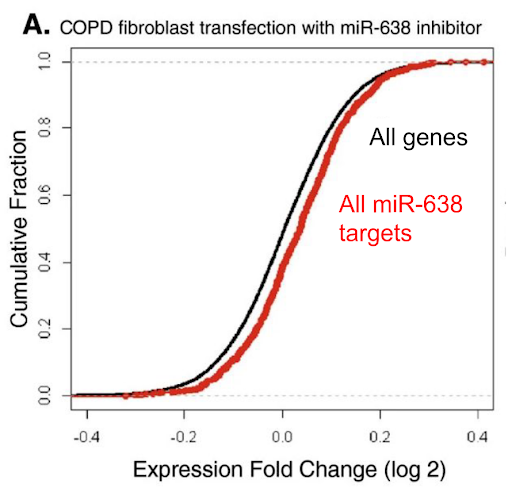
In this 2013 paper, Christenson et al. investigated the role of miR-638 microRNA in Chronic Obstructive Pulmonary Disease (COPD). The figure below is an ECDF plot that shows the effect of miR-638 inhibition on COPD fibroblast gene expression. Based on the plot, which of the following statement(s) is/are correct?
A) On average, miR-638 targets are upregulated in the transfected COPD fibroblasts compared to the set of all genes.
B) On average, miR-638 targets are downregulated in the transfected COPD macrophages compared to the set of all genes.
C) On average, miR-638 targets are downregulated in the transfected COPD fibroblasts compared to the set of all genes.
D) miR-638 activity downregulates target genes in COPD fibroblasts.
E) miR-638 activity upregulates target genes in COPD fibroblasts.
B3
You are analyzing genomic data from an organism with an unknown reference. Which of the following genome assembly strategies would be the most appropriate in this case?
A) De novo assembly using high-quality short reads only
B) De novo assembly using long or ultra-long reads only
C) Hybrid de novo assembly using short and long reads
D) Alignment to a closely related reference genome
E) A metagenomics-like assembly approach (utilizing a database of genomes)
B4
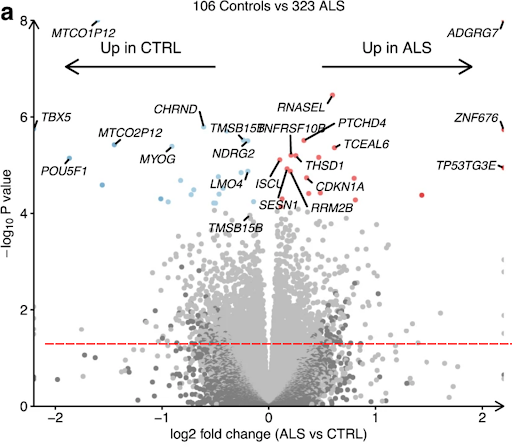
Ziff et al., 2023 describes the transcriptional landscape of Amyotrophic Lateral Sclerosis (ALS), a disease that causes motor neuron loss and often involves TDP-43 protein abnormalities. The analysis involved transcriptomic profiling of in vitro motor neuron samples generated using induced pluripotent stem cells derived from ALS patients and non-ALS controls (CTRL). Based on the volcano plot below, which of the following statements is/are false?
A) TCEAL6 expression is higher in CTRL
B) CDKN1A is up-regulated in ALS compared to CTRL
C) The p-value for CHRND differential gene expression is lower than that of MYOG
D) The p-value cutoff for differentially expressed genes in this plot is 4
E) The p-value cutoff for differentially expressed genes in this plot is 1.3
B5
To study the role of Dlx5 in mouse pup development, you used CRISPR-Cas9 technology to knock out that gene. CRISPR-Cas9 looked for a place in the genome that should be targeted, and Cas9 introduced a break. What are the primary DNA repair mechanisms the cell uses after CRISPR-Cas9’s activity, taking into account that it is a transcription factor that requires the 5'-TAATTA-3' consensus sequence for DNA binding?
A) base excision repair
B) mismatch repair
C) non-homologous recombination
D) microhomology-mediated end joining
Statistics
The second set of questions tested the participant’s familiarity with concepts in probability and statistics.
S1
Last year, 300 people applied for NGSchool. Their mean age was 27.2 (with a standard deviation of 4.5) years and their registration scores were normally distributed with a mean of 80 and a variance of 20. The easiest 6 questions were answered correctly by 100% participants and the hardest one was answered correctly by only one participant. Assuming that scoring is the same this year, what is the probability of receiving a score higher than 90? (*This is not the real participant data, a creative license was applied to design the above question.)
A) 0.500
B) 0.308
C) 0.022
D) 0.013
S2
While studying the effects of a new drug on immune cell function, you measured the proliferation rate of T-cells in the presence and absence of the drug. Which of the following statistical tests would be most appropriate for analyzing your data?
A) T-test
B) ANOVA
C) Chi-square test
D) Correlation analysis
S3
A researcher developed a logistic regression model that predicted the rate of lung fibrosis after chest radiotherapy based on patient sex, the percentage of lung tissue exposed to >20 Gy and single nucleotide polymorphisms in genes encoding for metalloproteinases. She validated the model in an independent population and found that sensitivity decreased from 98% to 94% and specificity decreased from 73% to 68% relative to performance on the initial study population. How can she assess model calibration in this scenario?
A) Using ROC curves
B) Based on the Akaike information criterion
C) By plotting the relation between the estimated risk and the observed proportion of events
D) By dividing the data into training, testing, and validation sets, but only using the first two during the model-building phase
S4
The prevalence of a metabolic disease in the population is 1 in 40,000. You designed a diagnostic method based on assessing the concentration of amino acid X in the spinal fluid using a cutoff value of 22 μmol/L. After running some tests, you determine that while using your new method 95% of tested healthy individuals receive a negative diagnosis, but 2% of people having the disease are incorrectly diagnosed.
Assuming your method were to be used as a mass screening tool across a hypothetical population of 2 million individuals (with general health metrics in line with national averages for metabolic markers and distribution of ages reflecting a typical urban population), calculate the approximate number of expected false positives.
A) 2000
B) 100 000
C) 40 000
D) 800
S5 - S6
Below are two discrete-time Markov chains with four states, represented by directed graphs. Take a look at the illustration and answer questions S5 and S6.
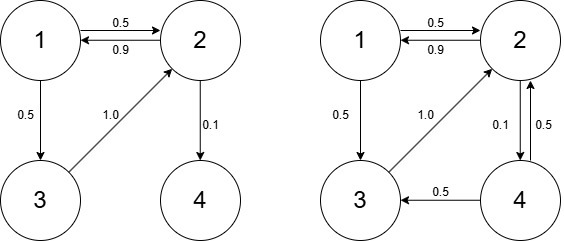
S5
Which of the following statement(s) is/are correct?
A) The Markov chain on the left has a steady state distribution
B) The Markov chain on the left has at least one absorbing state
C) The Markov chain on the left has at least one dispersive state
D) The Markov chain on the right has a steady state distribution
E) The Markov chain on the right has at least one absorbing state
F) The Markov chain on the right has at least one dispersive state
S6
For the Markov chain on the right, what is the probability that if we start in state 1, we return there in exactly three steps?
Solution: 0.45
S7
In many medical applications, it is of interest to analyze or model the time until an event takes place. A framework that is frequently used for this is survival analysis, where a survival function S(t) is related to a hazard rate h(t) by the relationship

The survival function describes the probability that the event of interest has happened before time t. Take a look at the below illustration of example survival functions (specifically their logarithm) as well as hazard rate functions, and answer the questions below.
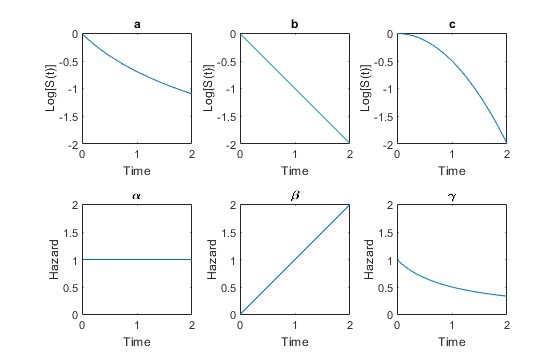
A) Which of the illustrated hazard rates (alpha, beta, and gamma) corresponds to each of the survival functions a, b and c? Note: please do not use special characters (⍺, β, 𝛾).
I) Hazard rate corresponding to a: (Solution: gamma)
II) Hazard rate corresponding to b: (Solution: alpha)
III) Hazard rate corresponding to c: (Solution: beta)
B) Looking at the hazard rates, which profile suits the moniker “memoryless” the best?
I) Alpha
II) Beta
III) Gamma
Coding
The last set of questions tested the basic coding skills of applicants to ensure they will be able to follow the practical part of the Summer School. While the practical part of the NGSchool program will be conducted using mainly Python and R, you could solve the following problems using your language or framework of choice.
C1
Given an input text file (input.tsv), parse the records to find a number made when the first digit and the last digit in each line is combined (in that order). What is the sum of all the numbers? Note that the same digit could be the first and the last digit in a line.
For example, if the lines are:
eightg1
4ninejfpd1jmmnnzjdtk5sjfttvgtdqspvmnhfbm
78seven8
6pcrrqgbzcspbdThe numbers for each line are 11, 45, 78 and 66 respectively. The sum of all the numbers would be 200.
Solution: 54950
Sample Code in R:
#importing the data
input <- read.table("input.tsv", quote="\"", comment.char="")
a1 <- strsplit(input$V1, split = "")
#Splitting each string into individual list of elements and extracting only numbers
x <- c()
for (i in 1:length(a1)) {
x[[i]] <- as.numeric(unlist(a1[[i]]))
x[[i]]<-x[[i]][!is.na(x[[i]])]
x[[i]] <- as.numeric(str_c(x[[i]], collapse = ""))
}
t1.num <- unlist(x)
t1.final <- c()
#Doing the summation based on criteria from question
for (i in 1:length(t1.num)) {
if (nchar(t1.num[[i]]) == 1) t1.final[[i]] <- as.numeric(paste0(t1.num[[i]], t1.num[[i]]))
else if (nchar(t1.num[[i]]) >= 2) t1.final[[i]] <- as.numeric(paste0(as.numeric(substr(t1.num[[i]], 1, 1)), t1.num[[i]] %% 10))
else t1.final[[i]] <- t1.num[[i]]
}
df <- unlist(t1.final)
colSums(data.frame(df))C2
Reading any number from left to right, if all the digits are in increasing order (for example 134468) we can call such a number an “increasing number”. Similarly, if all the digits are in decreasing order (for example 66420) we can call it a “decreasing number”. For this question, we shall call a positive integer that is neither increasing nor decreasing (for example 155349) a “bouncy number”. Clearly, there cannot be any bouncy numbers below one hundred, but just over half of the numbers below one thousand (525 exactly) are bouncy. In fact, the lowest number for which the proportion of bouncy numbers first reaches 50% is 538. Surprisingly, bouncy numbers become more and more common and by the time we reach 21780 the proportion of bouncy numbers is equal to 90%. Find the lowest number for which the proportion of bouncy numbers is exactly 99%.
Solution: 1587000
Sample Code in Python:
bouncyCount = 0
for i in range(1, 10000000):
increasing = list(str(i)) == sorted(list(str(i)))
decreasing = list(str(i)) == list(reversed(sorted(list(str(i)))))
bouncy = not increasing and not decreasing
if bouncy:
bouncyCount += 1
if bouncyCount/i == 0.99:
display(i)C3
The same DNA sequence template can produce different protein/peptide sequences as a result of different forward and reverse open-reading frames. Translate the below DNA sequence into all possible peptide sequences and answer the below questions.
Do not consider the start codon Methionine as part of the final peptide sequence.
>myseq
ATGGCACGTTTACGATCGTACTGAAGCGTACTGATGCGTACGATCGTACGTTTAACTG
ATGCGTAGCTGATGCGTTACTGACGTAGCGTAGTTTAGCGTAGCGTATGCTAACGCGT
ATCGTACGTTGATGCGTACTGATGCGTTTAGCGTACTGTAGCGTACTAGCGTACGTAAAHow many different peptide sequences can be created? Solution: 7
What is the length of the shortest peptide sequence? Solution: 2
What is the 2nd amino acid of the longest peptide? Provide a one-letter answer. Solution: L
Sample Code in R:
library(stringr)
library(bioseq)
#Input the DNA sequence
d<-c('ATGGCACGTTTACGATCGTACTGAAGCGTACTGATGCGTACGATCGTACGTTTAACTGATGCGTAGCTGATGCGTTACTG',
'ACGTAGCGTAGTTTAGCGTAGCGTATGCTAACGCGTATCGTACGTTGATGCGTACTGATGCGTTTAGCGTACTGTAGCGT', 'ACTAGCGTACGTAAA')
dna_seq <-paste0(d, collapse = '')
#Forward strand
dna_f <- dna(dna_seq, str_sub(dna_seq,2), str_sub(dna_seq,3))
# dna reverse
dna_r <- sapply(c(dna_seq, str_sub(dna_seq,2), str_sub(dna_seq,3)), function(x){
paste0(rev(strsplit(x, "")[[1]]), collapse = "")})
# dna complement
dna_r <- seq_complement(dna(dna_r))
aa_f <- seq_translate(dna_f)
aa_r <-seq_translate(dna_r)
get_peptides <- function(aa_seq){
sapply(aa_seq, function(aa){
idx <- as.numeric(gregexpr('M', aa)[[1]])
pep <- sapply(idx, function(i){
if (i == -1) return(NA) # no start codons
aa_trim <- str_sub(aa, i)
stop <- as.numeric(str_locate(aa_trim, '\\*')[1,1])
stop <- ifelse(is.na(stop), str_length(aa_trim), stop) # no stop codons
str_sub(aa_trim, 1, stop)
})
pep[!is.na(pep)] #empty peptide after triming
})
}
# peptide chains from forward
get_peptides(aa_f)
# peptide chains from reverse
get_peptides(aa_r)C4
The efficient parsing of biological databases is an essential skill for computational biologists. Use any approach, to retrieve information about the human gene located on chromosome 17, with genomic coordinates (in GRCh38.p13 reference genome) of chr17:60,149,942-60,179,021. This gene is translated to a protein involved in many pathological and physiological responses in human diseases. However, these processes are mostly studied using mouse models. Which of the below statement(s) is/are correct?
A) The human gene contains 10 exons
B) The human-mouse orthology type is “One to One”
C) The symbol for the mouse gene is also Ca4
D) The mouse gene and the human gene contain 8 exons each
E) The name for the corresponding human protein is CAH4
F) The protein transcribed in humans is working as a transcription factor